
Further progress! Chinese Academy of Medical Sciences
Li Ping ping's team reveals the mechanism of insulin receptor degradation in the liver


Insulin is a peptide hormone secreted mainly by the pancreatic islets of Langerhans and plays an important role in glucose homeostasis, cell growth, survival, proliferation, and β-cell metabolism. Insulin signaling is essential for glucose metabolism, and insulin reduces insulin receptor (InsR) levels in a dose-dependent and time-dependent manner. However, the mechanisms regulating the reduction of InsR after insulin stimulation are still poorly understood.
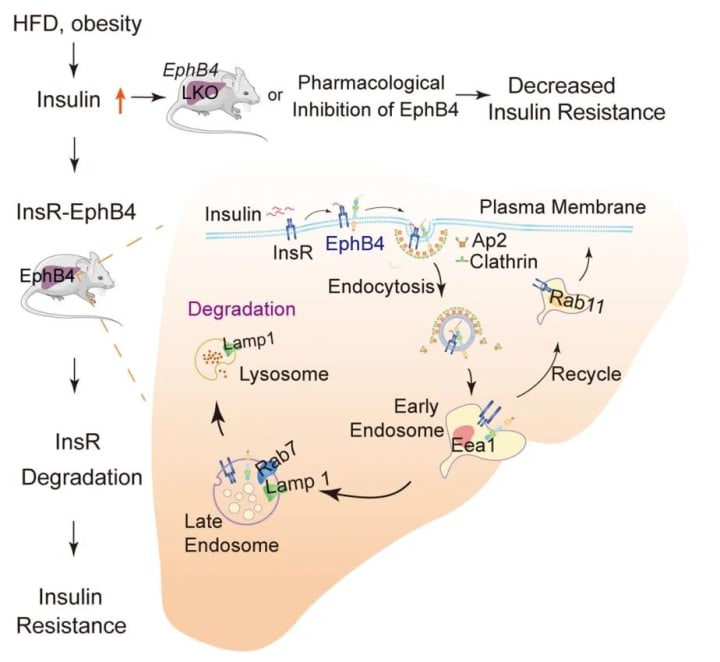
On September 21, 2022, Li Pingping's team at the Institute of Pharmaceutical Sciences, Peking Union Medical College, Chinese Academy of Medical Sciences, published a study entitled "Insulin induces insulin receptor degradation in the liver through EphB4". The study shows that EphB4 (Eph receptor B4), a tyrosine kinase receptor that regulates cell adhesion and migration, binds directly to InsR and that this interaction is significantly enhanced by insulin. The interaction between EphB4 and InsR facilitates lattice-protein-mediated endocytosis and degradation of InsR in lysosomes due to the complex binding motif of the adapter protein 2 (Ap2) in EphB4.
Overexpression of EphB4 in the liver reduced InsR levels and increased hepatic and systemic insulin resistance in food-fed mice, while genetic or pharmacological inhibition of EphB4 improved insulin resistance and glucose intolerance in obese mice. These results shed light on the role of EphB4 in insulin signaling and suggest that EphB4 may represent a therapeutic target for the treatment of insulin resistance and type 2 diabetes.
In addition, on August 5, 2022, Li Pingping from the Institute of Drug Research, Chinese Academy of Medical Sciences/Beijing Union Medical College, and Huang Zhifeng from Wenzhou Medical University co-authored a paper entitled "Hepatocyte Ltb4r1 promotes NAFLD development in obesity" published online in Hepatology (IF=17). The study found that hepatocyte-specific knockdown of Ltb4r1 improved hepatic steatosis and systemic insulin resistance in dietary and genetically induced obese mice. In Ltb4r1 HKO obese mice, mRNA levels of key enzymes involved in ab initio adipogenesis (DNL) and fatty acid esterification were reduced. LTB4/Ltb4r1 directly promoted adipogenesis in HepG2 cells and primary hepatocytes. Mechanistically, LTB4/Ltb4r1 promotes adipogenesis by activating the cAMP-PKA-IRE1α-XBP1s axis in hepatocytes, which in turn promotes the expression of adipogenic genes regulated by XBP1s. In addition, inhibition of Ltb4r1 by Ltb4r1 inhibitor or lentiviral-shRNA delivery attenuated the fatty liver phenotype in obese mice. In conclusion, this study found that LTB4/Ltb4r1 directly promotes adipogenesis in hepatocytes by activating PKA-IRE1α-XBP1s, thereby promoting the expression of adipogenic genes. Inhibition of hepatocyte Ltb4r1 improved hepatic steatosis and insulin resistance. Ltb4r1 is a potential therapeutic target for NAFLD (click to read).
On December 14, 2021, Huang Zhifeng from Wenzhou Medical University, Li Pingping from Peking Union Medical College, Chinese Academy of Medical Sciences, and Ren Jun from Zhongshan Hospital, Fudan University collaborated to publish an online article in Nature Communications titled "Paracrine FGFs target skeletal muscle to exert potent antioxidant activity". The study identified paracrine FGF4 as a potent anti-hyperglycemic FGF by testing multiple homologs of FGFR1c in parallel in diabetic mice. the study found that, like FGF1, paracrine FGF4 was also more effective than endocrine FGF21 in lowering blood glucose. In addition, this study found that paracrine FGF4 and FGF1 exert their superior glycemic control by targeting skeletal muscle, which expresses abundant FGFR1c but lacks β-klotho (KLB), an essential FGF21 co-receptor. Mechanistically, both FGF4 and FGF1 upregulate GLUT4 cell surface abundance in skeletal muscle in an AMPKα-dependent but non-insulin-dependent manner. rFGF4 chronic treatment ameliorates insulin resistance and inhibits adipose macrophage infiltration and inflammation. Notably, unlike FGF1 (a pan-FGFR ligand), FGF4 has a more limited FGFR1c binding specificity and has no significant effect on food intake. In conclusion, the potent anti-hyperglycemic and anti-inflammatory properties of FGF4 in this study demonstrate its potential application in the treatment of T2D and related metabolic disorders (click to read).
Insulin binds to the insulin receptor (InsR) and activates insulin signaling and downstream pathways, including PI3K-AKT, mTOR, and RAF1-MEK-ERK1/2 pathways, to mediate its metabolic effects. Impaired insulin action, called insulin resistance, accompanies obesity and leads to the development of type 2 diabetes and metabolic syndrome. Many in vivo studies have shown that insulin resistance is accompanied by hyperinsulinemia. In addition, hyperinsulinemia, especially in the basal state, has been shown to maintain, amplify, or trigger insulin resistance. For example, Ins transgenic mice have only a 2-fold increase in basal insulin concentrations and are characterized by severe insulin resistance.
In vivo studies have shown that insulin administration increases basal hyperinsulinemia to levels similar to those observed in the insulin-resistant state and has confirmed that basal insulin levels may contribute to insulin resistance. Several studies have shown that sustained exposure to high levels of insulin decreases InsR abundance in lymphocytes, adipocytes, and hepatocytes. This supports the idea that insulin itself perpetuates insulin resistance to protect cells from constitutive pathway activation. However, it is unclear whether this is a physiological event of insulin on its receptors.
This study found that EphB4 binds directly to InsR and mediates insulin-induced InsR degradation in hepatocytes and that obesity increased EphB4 expression in mouse and patient livers. epHB4 specifically binds to the intracellular structural domain of InsR, and insulin increased this interaction. the Ap2-binding motif in EPHB4 promotes InsR endocytosis and lysosomal degradation, and The Ap2 binding motif in EPHB4 promotes InsR endocytosis and lysosomal degradation and reduces InsR abundance and insulin sensitivity in the cell membrane. This suggests that EPHB4 acts as a co-receptor for InsR to inhibit insulin signaling and provides a molecular mechanism for insulin-induced degradation of hepatic InsR and systemic insulin resistance.
Further studies revealed that the EPHB4 kinase structural domain interacts with the most. In addition, binding motifs of Ap2 in EPHB4 are required for EPHB4-induced InsR degradation, and these binding motifs are present in the kinase structural domain of EPHB4. In addition, the kinase death mutant of EPHB4 (K647R) reduced the interaction of EPHB4 with INSR. These results suggest that the kinase structural domain and activity of EPHB4 are required for EPHB4-INSR interaction and INSR degradation.
Furthermore, it was found that EphB4 deficiency almost completely reversed the effect of insulin on InsR degradation in hepatocytes and that EPHB4 played a key role in this process.
A graphical summary of the effect of EphB4 on hepatic insulin receptor degradation (Figure from Nature Metabolism)
In conclusion, this study clearly shows that insulin decreases INSR protein levels in cell membranes and that insulin sensitivity is reduced during insulin treatment. Consistent with these results, after intensive insulin therapy, some diabetic patients often fail to improve glycemic control with further increases in insulin dose while increasing the risk of weight gain. Thus, this study reveals potential mechanisms for failure in glycemic management in some patients receiving insulin therapy.
About the Creator
Sue Torres
Is there any other reason to live to change the world?
Keep reading
More stories from Sue Torres and writers in Futurism and other communities.
MEAN Stack: 3 Use Cases that Make it a Go-To Choice
Introduction The MEAN Stack Application is an open-source collection of JavaScript technology that has earned significant popularity. Using MEAN stack components can help in building complex full-stack web applications, thus making it an excellent option for front and backend development.
By Mukesh Ram2 days ago in Futurism




Comments
There are no comments for this story
Be the first to respond and start the conversation.