
What Is The Role Of The Kidney In Blood Pressure Regulation?
Blood Pressure and Hypertension
The kidney plays a role in hypertension. A researcher published to date about 200 years ago is known that abnormalities in the production of urine by the kidney alter the blood in a way that increases vascular resistance, leading to high blood pressure. And the enlarged heart is mass. Several years later, Harry Goldblatt induced fatal hypertension in dogs by rupturing one of the renal arteries.
Arthur Guyton and his colleagues hypothesized that the kidney controls blood pressure levels in the 1970s by controlling the amount of extracellular fluid. They argued that the combination of dietary intake with urinary excreta of salt and water normally reached equilibrium, leading to a constant volume of single-cell fluid and blood pressure
They reported that when blood pressure increases for some reason, the perfusion pressure of the kidneys also increases, increasing the excretion of sodium and water, which Gitan called pressure natriuresis.
What Is The Role Of The Kidney In Blood Pressure Regulation?
Depending on the ability of the kidney to excrete sodium, this mechanism that alters blood pressure is of sufficient benefit to increase peripheral vascular resistance to limit the intravascular volume and, consequently, decrease blood pressure in response to a range of stimuli at frequencies. higher heart rates. It should be in addition, an allowable pressure modification - the natriuretic response presumably requires maintaining a chronic elevation of intra-arterial pressure, leading to a high equilibrium point in blood pressure for salt and water excretion. It moves
Furthermore, a series of kidney cross-transplant studies have supported an important role in the intrinsic functions of the kidney in the pathogenesis of hypertension. Genetically, compatible donor and recipient strains were used to avoiding rejection, with the elimination of both native kidneys so that the transplanted kidney provides full transplant function
Similarly, studies of spontaneous hypertensive mice and compatible hypertensive mice have rewritten these findings. The same principle is valid in humans where resistant hypertension can be reduced after a successful kidney transplant
Collectively, these studies point to the fact that decreased kidney excretion of sodium leads to hypertension.
Blood Pressure and Hypertension
Hypertension is one of the most common chronic diseases in humans, affecting more than one billion people worldwide.
Although high blood pressure generally does not cause obvious symptoms, the consequences of chronic hypertension, including cardiac hypertrophy, heart failure, stroke, and kidney disease, are responsible for considerable morbidity and mortality. Treatments that effectively lower blood pressure can prevent these complications
However, in recent times, blood pressure dropped to target blood levels in less than 50% of patients who received treatment for hypertension, and the rate was less than 40% in people with chronic kidney disease. (ERC)
The reasons for these poor results include health problems surrounding the patient's care, compliance and education processes. Furthermore, the exact cause of hypertension is unclear in the vast majority of hypertensive patients. Limitations in understanding the pathogenesis of hypertension in individual patients are a barrier to implementing personalized approaches to prevention and treatment and to identifying new and specific treatments.
How does the kidney increase blood volume?
Angiotensin-2 also stimulates the adrenal gland to secrete a hormone called aldosterone. Aldosterone further stimulates Na reabsorption in the distal tubule, and the water is recaptured with Na. Increased redistribution of Na and water from the distal tubule reduces urine output and increases the amount of circulating blood. Increasing the amount of blood helps the heart muscle dilate and this causes more pressure to be produced with each beat, which increases blood pressure. The amount of circulating blood is proportional to the stretching of the heart muscle.
Kidney actions to control blood pressure are particularly important during traumatic injury when they are necessary to maintain blood pressure and prevent fluid loss. The body stores calcium in the bones, but it also maintains constant levels of calcium in the blood. If calcium levels in the blood drop, the parathyroid glands in the neck release a hormone called parathyroid hormone. Parathyroid hormone increases calcium redevelopment of the duct outside the nephron to restore calcium levels in the blood. On the one hand, the parathyroid hormone also causes calcium absorption from the intestine by stimulating the release of calcium from the bone.
The body also requires vitamin D to stimulate calcium absorption from the kidneys and intestine. Vitamin D is found in dairy products. A precursor to vitamin D (cholecalciferol) is produced in the skin and processed in the liver. The final step in converting an inactive form of colylceferol to activated vitamin D occurs in the proximal duct of the nephron. Once activated, vitamin D stimulates the absorption of calcium from the proximal tubule and from the intestine, which increases calcium levels in the blood.
Kidney stones are abnormalities that are usually caused by problems in the kidney's ability to handle calcium. In addition, the role of the kidney in maintaining calcium in the blood is important in osteoporosis, a bone disease that affects many older people, especially women.
Therefore, the kidneys function in the body:
- Control blood composition and eliminate waste by filtration / reabsorption / secretion
- Blood pressure affected by renin secretion.
- Vitamin D helps regulate body calcium through activation.
If for some reason, the kidneys are not working, kidney dialysis methods (artificial filtration methods) become the only option to help the patient survive by cleaning the blood. This is especially necessary when both kidneys fail.
The Mechanisms of Blood Pressure are Controlled by the Kidneys.
1. Intrarenal actions of the renin-angiotensin system in the control of blood pressure
The renin-angiotensin system (RAS) is a powerful modulator of blood pressure, and the reduction of RAS produces hypertension. Drug blockade of RAS with renin inhibitors, angiotensin-converting enzyme (ACE) inhibitors, or angiotensin receptor blockers effectively reduces blood pressure in a substantial proportion of patients with hypertension - 10 /, such as human-caused RAS. Illustrates the critical role for activation. hypertension. In rodents, the removal of the RAS gene reduces blood pressure, causing hypertension to cause hypertension .
While the cells of the distal tubule (dense macula) do not feel Na in the filtrate either, and the arterial cells (juxtaglomerular cells) feel the blood pressure. Studies have shown that chronic infusion of a low dose of angiotensin II directly into the kidney caused hypertension with altered nature due to a change in the pressure-natriuresis relationship.
The existence of local and independent control of RAS activity within the kidney is also believed to affect sodium excretion and regulation of blood pressure. In this hypothesis, the increase in circulating levels of angiotensin II is associated with the accumulation of angiotensin peptides in the kidney, the primary expression of angiotensinogen in the proximal tubal epithelium, the primary substrate RAS and the increased excretion of angiotensinogen and peptide of angiotensin in the urine. In this pathway, angiotensin II through angiotensin type 1 (AT1) receptors in the kidney induce local activation of RAS within the kidney and increase the generation of angiotensin II in the lumen of the renal tubules, resulting in results in autocrine and paracrine stimulation occurs. Epithelial transporter.
In support of this view, recent studies have verified the critical need for ACE within the kidney, thus fully revealing the expression of the sodium transporter, renal sodium recombination, and the stimulation of hypertension in the context of RAS activation.
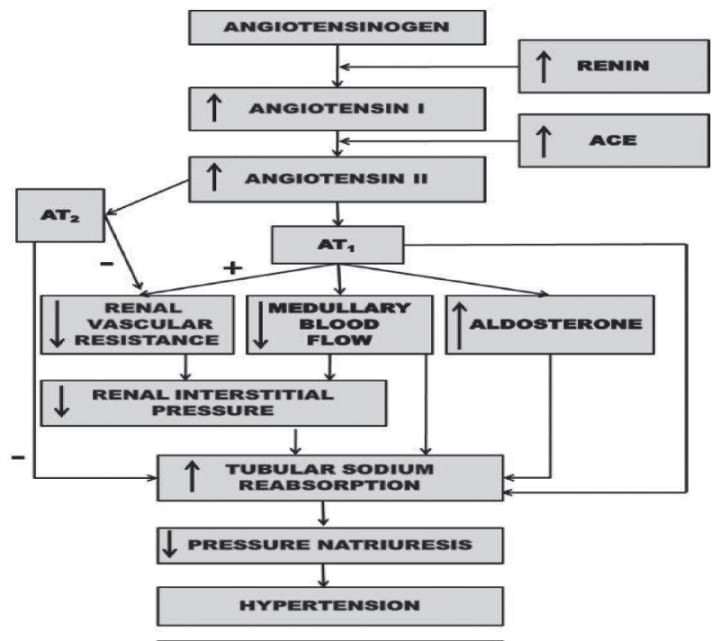
by heighpubs
Figure 1: Renal mechanism by which activation of the renin-angiotensin system reduces the ratio of pressure natriuresis and leads to hypertension .
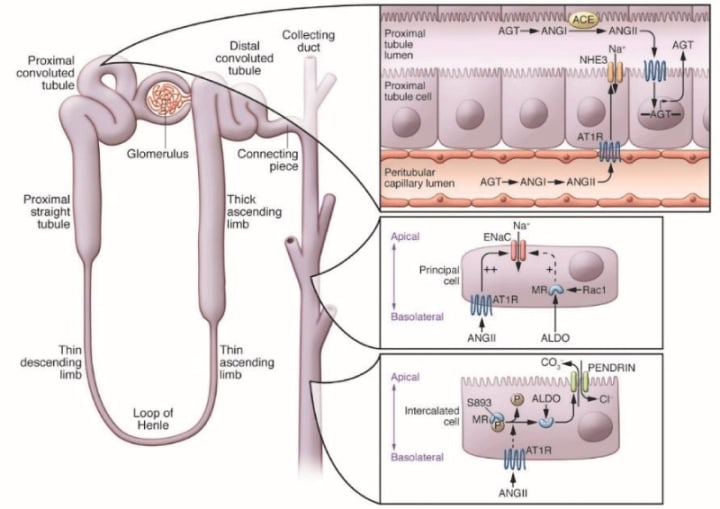
by heighpubs
Figure 2: A model for local control of RAS activity within the kidney: high levels of angiotensin II (ANGII) in the circulation, derived mainly from angiotensinogen (AGT) generated by the liver; Increased ANGII in the kidney, regulation of AGT in the epithelium of the proximal tubule, increased level of AGT in the tubular lumen, which requires ANGII requires expression of the angiotensin-converting enzyme (ACE) in the proximal tubule ( PT) and the edge of the brush. The excretion of AGT and ANG peptides increased in the urine .
2. New control mechanisms and sites for aldosterone in hypertension
AT1 receptors in the glomerulus zone of the adrenal gland stimulate the release of aldosterone, causing the downstream effect of aldosterone RAS. Activation of the mineralocorticoid receptor (MR) in aldosterone-sensitive nephron segments stimulates the assembly and translocation of ENAC subunits. Mutations in ENAC sub mutations that increase their degradation, resulting in membrane densities and open channel probability, are characterized by Liddle syndrome, which is similar to severe, early-onset hyperaldosteronism. Hypertension is characterized, but with low aldosterone levels. Similarly, activation mutations in the genes encoding MR also cause hypertension arising from changes in steroid hormones during pregnancy. These syndromes may highlight the potential for senescence of the MR / ENaC signalling pathway in the kidney to promote hypertension.
Aldosterone, in addition to stimulating sodium recombination, promotes the secretion of potassium in the urine. Shibata et al have shown in their study that MR phosphorylation regulates aldosterone responses in the kidney. They showed that phosphorylation of S843 in MR inhibits ligand binding. This form of MR is present only in the crossover cells of the collecting duct of the kidney, where its phosphorylation is differentially regulated by volume reduction and hyperkalemia. For example, in volume reduction, MR is dephosphorylated in interrelated cells, resulting in the multiplication of sodium and chloride recombination, allowing a different response to volume reduction. Although MR is classically activated by aldosterone, recent studies suggest that the small GTPase R1 may promote hypertension through an MRI-dependent pathway, even with aldosterone suppressed. Also, level (Figure 3).
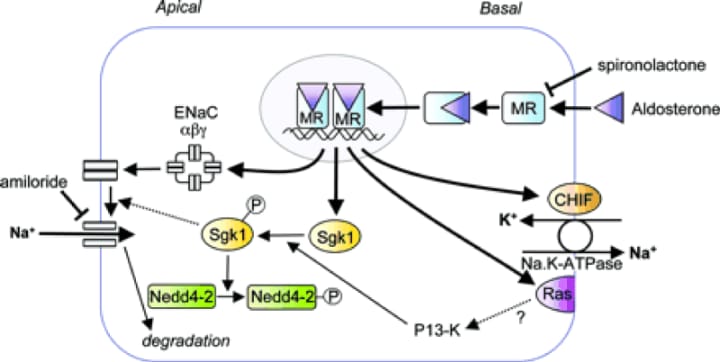
by heighpubs
Figure 3: Representation of an aldosterone-sensitive epithelial cell. The proteins encoded by aldosterone-induced genes are discussed in the text: ENAC α, β and,, CHIEF, sgk and RAS are indicated which are their known or therapeutic functions.
3. WNK: new routes that regulate the renewal of solute transport
Reliable evidence showing an important role for the kidney in blood pressure regulation has defined the genetic basis for almost all Mendelian disorders associated with abnormal blood pressure phenotypes in humans. In each case, these mutations affect the recombination of sodium and liquid with nephrons. One of these disorders is type II pseudohypoaldosteronism (PHAII), a Mendelian syndrome characterized by an unusual combination of hypertension and hyperkalemia, caused by a mutation in a gene encoding the WNK1 kinase (without lysine [K]). goes. WNK4. This discovery accelerated the in-depth study of these unique eunuchs, thus identifying the roles of WNK1 and WNK4 in regulating sodium and potassium flow in the distal nephron. These actions are mainly mediated by the control of the relative levels and activities of thiazide-sensitive sodium chloride (Na) cotransporters (NCC) and/or renal channels of external medullary potassium (K) (ROMK, NCC represents a major pathway for sodium recombination in the distal nephron and is a target for thiazide diuretics, which are effective and widely used antihypertensive agents. A cornerstone of treatment for thiazide PHAII., Which is consistent with the findings that NCC is an important feature of overactivity disorder. It is worth noting that while the actions of WNK4 to suppress ROMK activity have been consistently reported in these studies, NCC activity is Variable effects of WNK4 have been observed, probably related to relative levels of WNK4 in experimental systems. In this regard, the accumulation of endogenous WNK4 leads to N20 mutation activity, possibly STE20 / SPS-1-related proline-alanine-containing protein kinase (SPAK). ) Through phosphorylation, whereas WNK4 De K appears to clearly target NCC for lysosomal degradation of overgrowth. (Fig. 4).
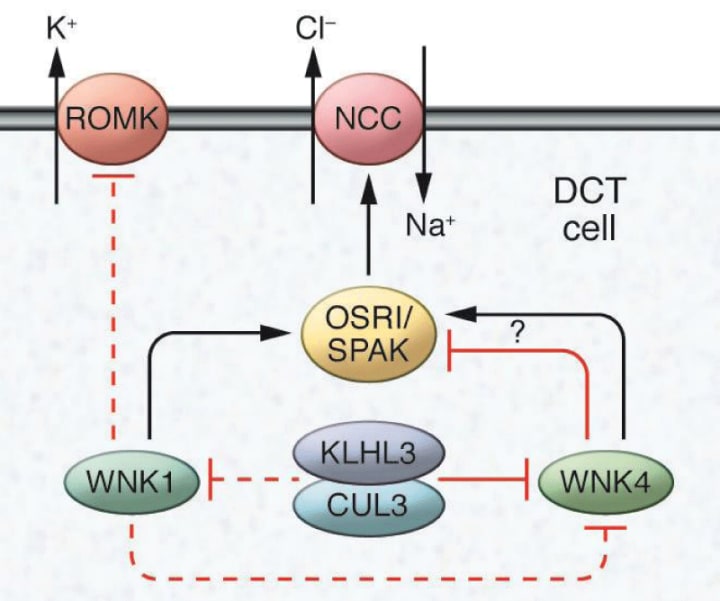
Figure 4: Mechanisms that regulate sodium and potassium flow in distal nephrons
The WNK family sodium chloride cotransporters (NCC) and the kidneys regulate the activity of the external distal potassium channel (ROMK) in the cells of the distal convulsive tubule (DCT) in the kidney. WNK1 phosphorylates and stimulates SPS1-related proline / alanine-rich protein kinase (SPAK) and stress-reactive oxidative protein kinase 1 (OSR1), which in turn promotes NCC-dependent sodium transport. WNK1 can also inhibit ROMK. WNK4 inhibits ROMK, but both excitatory and inhibitory actions in NCC have been reported to occur according to the experimental system used. WNK4 levels are regulated by the activity of cullin 3-KLHL3 ubiquitin ligase, which has also been suggested to modulate WNK1.
4. How the flow of sodium and potassium in the distal nephron is controlled.
Increased NCC activity through WNK modulation is a final common pathway for the development of hypertension in many settings. For example, adrenergic stimulation of blood increases blood pressure by suppressing WNK4 and, in turn, increases NCC activity. Furthermore, calcineurin inhibitors are commonly used to treat autoimmune diseases and prevent transplant rejection, which often causes hypertension. Recent studies by Ellison et al have indicated that the hypertension mechanism associated with the use of calcineurin inhibitors involves the stimulation of NCC through the degradation of WNK3.
While the continued delineation of WNK functions has provided important information about renal physiology, only a small subset of patients with PHAII has a mutation in the WNK gene. Using exome sequencing, the Lifton group mutates the Kelch3 (KLHL3) and Cullin3 (CUL3) genes in patients with PHAII . Furthermore, mutations in both genes cause disease in approximately 80% of individuals affected by PHAII . KLHL3 is one of a family of more than 50 Kelch proteins that contain wide-width complex Bric-a-Brax (BTB-containing) complexes, tram track, characterized by a β-helix domain of six bleeds to bind specific target proteins. is. Provides scaffolding for the CUL3 complex, which includes BTB domain proteins like KLHL3 and a RING domain protein that acts as an E3 ubiquitin ligase, targeting protein substrates specific for ubiquitylation (Fig. 5)
by heighpubs
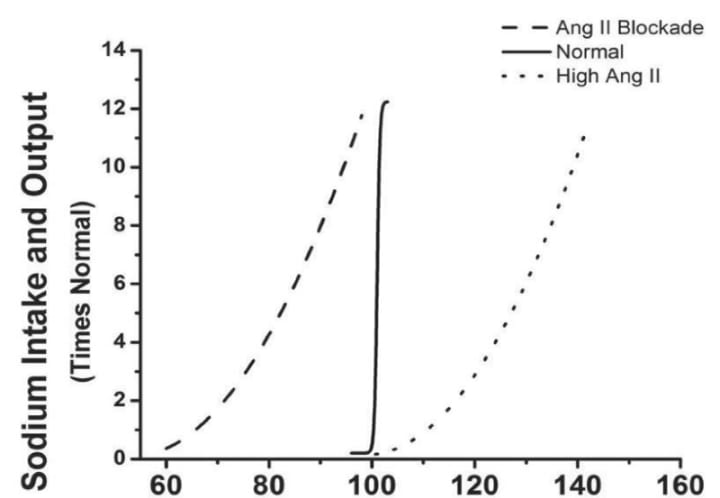
Figure 5: Effect of changes in the middle artery during chronic changes in sodium intake after angiotensin-converting enzyme (ACE) inhibition, or when angiotensin II inhibits angiotensin II at a consistently low dose (5 ng / kg/min). It was infected by. Sodium intake increased when it was suppressed. (Redfern of data in Hall et al, 1980) .
5. Homeostasis of salt
Salt sensitivity, defined as exaggerated changes in blood pressure in response to extremes in diet sensitivity, is relatively common and is associated with an increased risk of developing hypertension. Classic Gaitonian models suggest that reductions in the excretion of sodium by the kidneys are the basis of sensitivity to salt, with altered sodium elimination during high-salt feeding that leads directly to the increased volume of extracellular fluid, which promotes an increase in blood pressure. This model assumes that the two main components of the extracellular volume are in balance within the intravascular and interstitial spaces. Therefore, the accumulation of sodium will be accompanied by greater retention of water to maintain iso-osmolality and this will increase the proportional volume.
However, the study by Titz et al. Recently it has been indicated that sodium management is more complex than this classic two-compartment model; The interstitium of the skin can act as sodium reserves, affecting the effect of sodium accumulation on intravascular volume and blood pressure. During high-salt feeding, sodium freezes in the subdermal interstitium at hypertonic concentrations in complexes with protroglucenes . Macrophages infiltrating the interstitial space detect hypertonicity caused by this accumulation of sodium in excess water, triggering the expression of TonOEBP, a transcription factor that regulates the expression of osmoprotective genes. One of the genes induced downstream of TONEBP is vascular endothelial growth factor C (VEGF-C), which is a potent indicator of lymphatic angiogenesis.
In response to high-salt feeding, Titz's group found strong lymphatic vessel hyperplasia in the dermal interstate. Macrophage depletion, specific removal of TONEBP cells from macrophages, or specific VEGAF-C blockade prevented hyperplasia of lymphatic vessels and increased the level of sodium-dependent hypertension, indicating that this way has a strange presence. An important role is the control of the volume of sodium and liquid. Elevated plasma levels of VEGF-C were observed in patients with refractory hypertension, indicating that this system may be altered in the human disorder. However, preclinical models predict that decreased VEGF-C levels will promote hypertension. However, chronic hypertension in humans is a complex disorder; It is possible that the observed absorption at VEGAF-C levels reflects tissue resistance to VEGAF-C or even compensatory responses.
6. Hypertensive kidney injury and the progression of chronic kidney disease.
The kidney remains an important site for hypertensive organ loss, ranking second as the leading cause of End-Stage Renal Disease (ESRD) for diabetic nephropathy. Furthermore, the presence of chronic kidney disease (CKD), including hypertension, has been shown to be a strong independent risk factor for adverse cardiovascular outcomes. However, key aspects of clinical hypertensive kidney disease are poorly understood, such as marked differences in individual susceptibility to hypertensive kidney damage and categorical variable renoprotective effects of classes of antihypertensive drugs.
Studies have shown that time-varied SBP was associated with incident CKD, with a constant increase in incident risk CKD above SBP of 120 mmHg. Time-weighted SBP was associated with a more rapid decrease in kidney function. Diabetes was the strongest predictor of CKD, and a rapid decline in kidney function and increased glycemic control were associated with increased risk, supporting the role of BP and other traditional risk factors, such as progression of renal function in diabetes and onset and Hypertension decreases in patients with normal renal function at baseline.
Note:
Kidney sodium management is an important determinant of intra- and extra-renal blood pressure levels, and is under complex physiological control by hormones, inflammatory mediators, and the sympathetic nervous system. Clearly, a basic mechanism of efficacy for diuretics and dietary sodium restriction in hypertension is to favorably affect sodium balance and homeostasis. Other antihypertensive agents such as RAS inhibitors, vasodilators, and block blockers work through a similar mechanism by facilitating pressure natrioresis. Recent studies have also suggested that pathways that regulate WNK signaling pathways, soluble inflammatory mediators, and extrarenal disposition of sodium may also be useful targets for increasing sodium clearance and lowering blood pressure in hypertension.
The renin-angiotensin system (RAS) is a powerful modulator of blood pressure, and RAS dysfunction causes hypertension. Drug blockade of RAS with renin inhibitors, angiotensin-converting enzyme (ACE) inhibitors, or angiotensin receptor blockers effectively reduces blood pressure in a substantial proportion of patients with hypertension - 10 /, such as human-caused RAS. Illustrates the critical role for activation. hypertension. Similarly, in rodent models, deletion of the RAS gene reduces blood pressure, while hypertension causes hypertension.
End Conclusion
There is an essential relationship between kidney control and blood pressure. An attenuated ability of the kidney to excrete sodium in response to hypertension is a major contributor to hypertension, even if the onset is the cause. In this sense, new pathways that regulate key sodium transporters in renal epithelia have an important effect on the pathogenesis of hypertension, supporting a model in which altered renal excretion of sodium is the last common pathway through which vessels, nerves and nerves. Inflammatory reactions increase blood pressure. The relationship between sodium intake and changes in body fluid volume reveal mechanisms.
That's it for the article, like , comment and share with your family. If you really like my work then you can send me a tip as a gift so that I can provide you such content.
About the Creator
Mustafa Rangoonwala
Hello Reader,
My Name is Mustafa Rangoonwala, I am an Holistic Practitioner since last 7+ Years. I am a Graphologist, NLPMP, Reiki Master Practitioner, Ganotherapist and Vastu Consultant.
I have recently joined Vocal..Support me !!!!!!
Keep reading
More stories from Mustafa Rangoonwala and writers in FYI and other communities.
Can You Get Your Period And Still Be Pregnant In The First Month
If there is something that characterizes pregnancy, it is the absence of menstruation, at least until the quarantine has passed after having given birth. In fact, many women find that they may be pregnant after realizing they have a missed or late period, without ever noticing any other symptoms.
By Mustafa Rangoonwala3 years ago in FYI
The Bloody Blood in Your Veins
Our Origins Today, I read something I didn't know—one more thing to add to all the other things I didn't know. By the time I grow up, I’ll have a collection. But it was intriguing, and it inspired me to write this post. I hope you enjoy it.
By Rene Volpi 3 days ago in FYI
What is the importance of crocodiles in the planet?
"Crocodile" The living creature that looks like lazy and slow but when any other creature whether that's an animal or human comes in their territory, they never compromise hunting them. The real power of silence can be determined from crocodile's nature.
By Parveen Baloch 4 days ago in FYI




Comments
There are no comments for this story
Be the first to respond and start the conversation.